干旱区研究 ›› 2022, Vol. 39 ›› Issue (5): 1618-1630.doi: 10.13866/j.azr.2022.05.26 cstr: 32277.14.AZR.20220526
牟红霞1( ),刘秉儒2(),李子豪2,李国旗1,麻冬梅1
),刘秉儒2(),李子豪2,李国旗1,麻冬梅1
收稿日期:2022-01-24
修回日期:2022-04-22
出版日期:2022-09-15
发布日期:2022-10-25
作者简介:牟红霞(1996-),女,硕士研究生,主要从事植物生态学研究. E-mail: 基金资助:
MOU Hongxia1(),LIU Bingru2(),LI Zihao2,LI Guoqi1,MA Dongmei1
Received:2022-01-24
Revised:2022-04-22
Published:2022-09-15
Online:2022-10-25
摘要:
为探究矿井水排放对荒漠草原土壤微生物群落结构及多样性的影响,以宁夏干旱风沙区矿井水排放地的荒漠草原为研究对象,通过高通量测序分析矿井水排放湖泊沿岸水滨区域(MJTA)、近岸陆域(MJTB)和自然区域(MJTC)土壤中0~10 cm、10~20 cm和20~30 cm土层中细菌和真菌群落组成,结合土壤理化因子等生境因素,揭示矿井水干扰对土壤微生物群落结构组成及多样性影响的主要因素。结果表明:(1) 矿井水排放显著改变了土壤细菌和真菌的群落组成,对土壤细菌和真菌群落门水平相对丰度差异影响显著。(2) 水滨区域、近岸陆域和自然区域中主要优势细菌门均为放线菌门(Actinobacteria)和变形菌门(Proteobacteria),主要优势细菌属均为: norank_f__norank_o__norank_c__MB-A2-108;水滨区域、近岸陆域和自然区域中主要优势真菌门均为子囊菌门(Ascomycota),水滨区域主要优势真菌属为unclassified_c__Sordariomycetes,近岸陆域和自然区域主要优势真菌属为光黑壳属(Preussia)。(3) 矿井水外排导致荒漠草原水滨区域土壤盐分、水分显著高于近岸陆域和自然区域,盐分抑制了土壤细菌和真菌的生长,但同时促进了研究区嗜盐碱细菌的富集。(4) 水滨区域土壤细菌和真菌多样性与丰富度均显著低于自然区域,进一步说明矿井水对土壤细菌和真菌丰富度与多样性影响显著;不同土层土壤细菌和真菌的丰富度与多样性差异较小;土壤环境因子与土壤细菌和真菌多样性分析表明土壤盐分、含水量、有机碳和pH是影响荒漠草原土壤细菌和真菌群落多样性的主要影响因子。
牟红霞,刘秉儒,李子豪,李国旗,麻冬梅. 矿井水对荒漠草原土壤微生物群落结构及多样性的影响[J]. 干旱区研究, 2022, 39(5): 1618-1630.
MOU Hongxia,LIU Bingru,LI Zihao,LI Guoqi,MA Dongmei. Effects of mine water on soil microbial community structure and diversity in desert steppe[J]. Arid Zone Research, 2022, 39(5): 1618-1630.
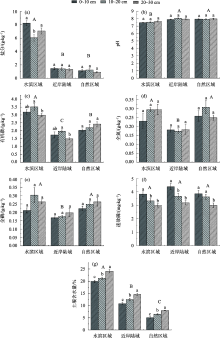
图1
不同区域不同深度土壤理化指标 注:图中数据为平均值±标准误;不同小写字母代表不同土层深度,不同大写字母表示不同区域,在P<0.05水平下差异显著。下同。"


图2
土壤细菌和真菌的NMDS分析 注:MJTA表示水滨区域;MJB表示近岸陆域;MJTC表示自然区域。下同。"

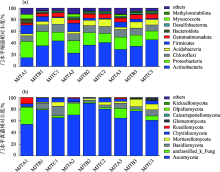
图3
土壤细菌和真菌门水平相对丰度"


图4
不同区域细菌和真菌门水平丰度差异"

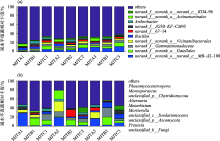
图5
土壤细菌和真菌属水平相对丰度"


图6
不同区域不同深度土壤细菌和真菌多样性指数"


图7
土壤细菌和真菌群落门水平与环境因子相关性分析 注:salt 表示土壤盐分;pH 表示土壤pH;SOC 表示土壤有机碳;TN 表示土壤全氮;TP 表示土壤全磷;AP 表示土壤速效磷;SM 表示土壤含水量。下同。"


图8
土壤细菌和真菌群落-CCA分析"

| [1] |
Manceau A, Merkulova M, Murdzek M, et al. Chemical forms of mercury in pyrite: Implications for predicting mercury releases in acid mine drainage settings[J]. Environmental Science and Technology, 2018, 52(18): 10286-10296.
doi: 10.1021/acs.est.8b02027 pmid: 30169032 |
| [2] |
Mcdevitt B, Cavazza M, Beam R, et al. Maximum removal efficiency of barium, strontium, radium, and sulfate with optimum AMD-Marcellus flowback mixing ratios for beneficial use in the northern Appalachian Basin[J]. Environmental Science and Technology, 2020, 54(8): 4829-4839.
doi: 10.1021/acs.est.9b07072 pmid: 32250106 |
| [3] | Deng J, Bai X, Zhou Y, et al. Variations of soil microbial communities accompanied by different vegetation restoration in an open-cut iron mining area[J]. Science of the Total Environment, 2020, 20(704): 135243. |
| [4] |
Cruz-Paredes C, Wallander H, Kjøller R, et al. Using community trait-distributions to assign microbial responses to pH changes and Cd in forest soils treated with wood ash[J]. Soil Biology and Biochemistry, 2017, 112: 153-164.
doi: 10.1016/j.soilbio.2017.05.004 |
| [5] |
Steven B, Gallegos-Graves L V, Yeager C M, et al. Dryland biological soil crust cyanobacteria show unexpected decreases in abundance under long-term elevated CO2[J]. Environmental Microbiology, 2012, 14(12): 3247-3258.
doi: 10.1111/1462-2920.12011 pmid: 23116182 |
| [6] |
Guo A, Zhao Z, Zhang P, et al. Linkage between soil nutrient and microbial characteristic in an opencast mine, China[J]. Science of the Total Environment, 2019, 671: 905-913.
doi: 10.1016/j.scitotenv.2019.03.065 |
| [7] |
Yao H, He Z L, Wilson M J, et al. Microbial biomass and community structure in a sequence of soils with increasing fertility and changing land use[J]. Microbial Ecology, 2000, 40(3): 223-237.
pmid: 11080380 |
| [8] | 安韶山, 李国辉, 陈利顶. 宁南山区典型植物根际与非根际土壤微生物功能多样性[J]. 生态学报, 2011, 31(18): 5225-5234. |
| [An Shaoshan, Li Guohui, Chen Liding. Soil microbial functional diversity between rhizosphere and non-rhizosphere of typical plants in the hilly area of southern Ningxia[J]. Acta Ecologica Sinica, 2011, 31(18): 5225-5234. ] | |
| [9] |
Shen Y, Chen W, Yang G, et al. Can litter addition mediate plant productivity responses to increased precipitation and nitrogen deposition in a typical steppe?[J]. Ecological Research, 2016, 31(4): 579-587.
doi: 10.1007/s11284-016-1368-5 |
| [10] |
Newman C, Agioutantis Z, Leon G B J. Assessment of potential impacts to surface and subsurface water bodies due to longwall mining[J]. International Journal of Mining Science and Technology, 2017, 27(1): 57-64.
doi: 10.1016/j.ijmst.2016.11.016 |
| [11] | Valkanas M M, Trun N J. A seasonal study of a passive abandoned coalmine drainage remediation system reveals three distinct zones of contaminant levels and microbial communities[J]. MicrobiologyOpen, 2018, 7(4): e00585. |
| [12] |
He J W, Li W X, Liu J, et al. Investigation of mineralogical and bacteria diversity in Nanxi River affected by acid mine drainage from the closed coal mine: Implications for characterizing natural attenuation process-ScienceDirect[J]. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, 2019, 217: 263-270.
doi: 10.1016/j.saa.2019.03.069 |
| [13] |
Sun S Y, Sun H, Zhang D S, et al. Response of soil microbes to vegetation restoration in coal mining subsidence areas at Huaibei Coal Mine, China[J]. International Journal of Environmental Research and Public Health, 2019, 16(10): 1757.
doi: 10.3390/ijerph16101757 |
| [14] | 陈迪. 高硫煤废弃矿井微生物群落演替规律及铁硫代谢基因的功能预测[D]. 北京: 中国矿业大学, 2020. |
| [Chen Di. Succession Rule of Microbial Communities and Function Prediction of Iron-sulfur Sulfur Metabolic Genes in High-sulfur Abandoned Coal Mines[D]. Beijing: China University of Mining and Technology, 2020. ] | |
| [15] | 李启艳, 翁炳霖, 李宗勋, 等. 稀土矿废弃地植被恢复过程中土壤微生物演变[J]. 中国环境科学, 2019, 39(10): 4360-4368. |
| [Li Qiyan, Weng Binglin, Li Zongxun, et al. Soil physicochemical characteristics and microbial evolution during vegetation restoration in ionic rare earth ore heap leaching waste land[J]. China Environmental Science, 2019, 39(10): 4360-4368. ] | |
| [16] | 彭玙萍, 曾伟民. 紫金山铜矿酸性矿山废水微生物群落多样性[J]. 微生物学通报, 2020, 47(9): 2887-2896. |
| [Peng Yuping, ZengWeimin. Diversity of microbial community in acid mine drainage from Zijinshan copper mine[J]. Microbiology Bulletin, 2020, 47(9): 2887-2896. ] | |
| [17] | 于小娟. 矿废水排放湖泊沿岸土壤性质及AM真菌空间分布特征的研究[D]. 杨凌: 西北农林科技大学, 2020. |
| [Yu Xiaojuan. Study on the Soil Properties and the Spatial Distribution Characteristics of Am Fungi along the Shore of the Mine Water Discharge Lake[D]. Yangling: Northwest A & F University, 2020. ] | |
| [18] | 鲍士旦. 土壤农化分析[M]. 北京: 中国农业出版社, 2000. |
| [Bao Shidan. Soil Agrochemical Analysis[M]. Beijing: China Agriculture Press, 2000. ] | |
| [19] |
Xu L, He N, Li X, et al. Local community assembly processes shape β-diversity of soil phoD-harbouring communities in the Northern Hemisphere steppes[J]. Global Ecology and Biogeography, 2021, 30(11): 2273-285.
doi: 10.1111/geb.13385 |
| [20] |
Magoč T, Salzberg S L. Flash: fast length adjustment of short reads to improve genome assemblies[J]. Bioinformatics, 2011, 27(21): 2957-2963.
doi: 10.1093/bioinformatics/btr507 pmid: 21903629 |
| [21] |
Edgar R C. UPARSE: highly accurate OTU sequences from microbial amplicon reads[J]. Nature Methods, 2013, 10(10): 996-998.
doi: 10.1038/nmeth.2604 pmid: 23955772 |
| [22] |
Stackebrandt E, GOEBEL B M. Taxonomic note: a place for DNA-DNA reassociation and 16S rRNA sequence analysis in the present species definition in bacteriology[J]. International Journal of Systematic and Evolutionary Microbiology, 1994, 44(4): 846-849.
doi: 10.1099/00207713-44-4-846 |
| [23] | 孙建平, 刘雅辉, 左永梅, 等. 盐地碱蓬根际土壤细菌群落结构及其功能[J]. 中国生态农业学报, 2020, 28(10): 1618-1629. |
| [Sun Jianping, Liu Yahui, Zuo Yongmei, et al. The bacterial community structure and function of Suaeda salsa rhizosphere soil[J]. Chinese Journal of Ecological Agriculture, 2020, 28(10): 1618-1629. ] | |
| [24] | 程丽芬, 樊兰英, 张欣, 等. 山西左云县采煤区人工湿地冬季沉积物细菌群落多样性[J]. 微生物学通报, 2019, 46(12): 3181-3192. |
| [Cheng Lifen, Fan Lanying, Zhang Xin, et al. Diversity of microbial community in winter at constructed wetlands sediment around coal mining area at Zuoyun, Shanxi Province[J]. Microbiology Bulletin, 2019, 46(12): 3181-3192. ] | |
| [25] | 牛世全, 龙洋, 李海云, 等. 应用IlluminaMiSeq高通量测序技术分析河西走廊地区盐碱土壤微生物多样性[J]. 微生物学通报, 2017, 44(9): 2067-2078. |
| [Niu Shiquan, Long Yang, Li Haiyun, et al. Microbial diversity in saline alkali soil from Hexi Corridor analyzed by Illumina MiSeq high-throughput sequencing system[J] Bulletin of Microbiology, 2017, 44(9): 2067-2078. ] | |
| [26] |
Ma B, Gong J. A meta-analysis of the publicly available bacterial and archaeal sequence diversity in saline soils[J]. World Journal of Microbiology and Biotechnology, 2013, 29(12): 2325-2334.
doi: 10.1007/s11274-013-1399-9 pmid: 23756871 |
| [27] | Canfora L, Bacci G, Pinzari F, et al. Salinity and bacterial diversity: to what extent does the concentration of salt affect the bacterial community in a saline soil?[J]. PLoS One, 2014, 9(9): e106662. |
| [28] | 李明, 毕江涛, 王静. 宁夏不同地区盐碱化土壤细菌群落多样性分布特征及其影响因子[J]. 生态学报, 2020, 40(4): 1316-1330. |
| [Li Ming, Bi Jiangtao, Wang Jing. Bacterial community structure and key influencing factors in saline soils of sites regions Ningxia[J]. Chinese Journal of Ecology, 2020, 40(4): 1316-1330. ] | |
| [29] |
Chen C, Xu X J, Xie P, et al. Pyrosequencing reveals microbial community dynamics in integrated simultaneous desulfurization and denitrification process at different influent nitrate concentrations[J]. Chemosphere, 2017, 171: 294-301.
doi: S0045-6535(16)31705-2 pmid: 28027473 |
| [30] | 李彬, 杨爱江, 胡霞, 等. 锑矿废水影响下水库沉积物中细菌群落结构特征研究[J]. 微生物学通报, 2021, 48(9): 2956-2971. |
| [Li Bin, Yang Aijiang, Hu Xia, et al. Bacterial community structure in reservoir sediments under the influence of antimony ore waste water[J]. Bulletin of Microbiology, 2021, 48(9): 2956-297. ] | |
| [31] | 杨珊珊, 张晓波, 陈邬锦, 等. 新疆三个不同盐碱地区土壤沉积物中细菌多样性分析[J]. 生物资源, 2021, 43(5): 453-460. |
| [Yang Shanshan, Zhang Xiaobo, Chen Wujin, et al. Analysis of bacterial diversity in soil sediments of three saline alkali areas in Xinjiang[J]. Biological Resources, 2021, 43(5): 453-460. ] | |
| [32] | 曹子敏, Joseph Frazer Banda, 裴理鑫, 等. 安徽某铁矿不同矿山废水库中微生物群落结构特征[J]. 微生物学报, 2019, 59(6): 1076-1088. |
| [Cao Zimin, Joseph Frazer Banda, Pei Lixin, et al. Microbial community structure characteristics in different mine drainage lakes of an iron mine in Anhui Province[J]. Journal of Microbiology, 2019, 59(6): 1076-1088. ] | |
| [33] |
单爱琴, 张燕婷, 肖洁, 等. 废弃矿井微生物群落演替特征实验研究[J]. 环境科学与技术, 2019, 42(4): 31-37.
doi: 10.1021/es072039a |
|
[Shan Aiqin, Zhang Yanting, Xiao Jie, et al. Experimental study of microbial community succession characteristics in abandoned mine groundwater[J]. Environmental Science and Technology, 2019, 42(4): 31-37. ]
doi: 10.1021/es072039a |
|
| [34] | 李善家, 王福祥, 从文倩, 等. 河西走廊荒漠土壤微生物群落结构及环境响应[J/OL]. 土壤学报, 2022. http://kns.cnki.net/kcms/detail/32.1119.P.20220307.1015.002.html. |
| [Li Shanjia, Wang Fuxiang, Cong Wenqian, et al. Microbial community structure and environmental response of desert soil in Hexi Corridor[J/OL]. Acta Pedologica Sinica, 2022. http://kns.cnki.net/kcms/detail/32.1119.P.20220307.1015.002.html. ] | |
| [35] | Zhao X, Liu H L, Yang P, et al. Effects of drip irrigation on bacterial diversity and community structure in rhizosphere soil of alfalfa[J]. Microbiology China, 2019, 46(10): 2579-2590. |
| [36] | 王海英, 郭守玉, 黄满荣, 等. 子囊菌较担子菌具有更快的进化速率和更高的物种多样性[J]. 中国科学: 生命科学, 2010, 40(8): 731-737, 765-772. |
| [Wang Haiying, Guo Shouyu, Huang Manrong, et al. Ascomycetes have a faster evolution rate and higher species diversity than Basidiomycetes[J]. China Science: Life Science, 2010, 40(8): 731-737, 765-772. ] | |
| [37] | 颜培, 杜远达, 姜爱霞, 等. 黄河三角洲土壤真菌群落结构及互作网络对盐度的响应[J]. 分子植物育种, 2021, 19(11): 3818-3828. |
| [Yan Pei, Du Yuanda, Jiang Aixia, et al. Response of soil fungal community structures and interaction networks to salinity in the Yellow River Delta[J]. Molecular Plant Breeding, 2021, 19(11): 3818-3828. ] | |
| [38] | 王艳云, 郭笃发. 黄河三角洲盐碱地土壤真菌多样性[J]. 北方园艺, 2016(18): 185-189. |
| [Wang Yanyun, Guo Dufa. fungal Diversity of saline alkali soil in Yellow River Delta[J]. Northern Horticulture, 2016(18): 185-189. ] | |
| [39] | 何苑皞, 周国英, 王圣洁, 等. 杉木人工林土壤真菌遗传多样性[J]. 生态学报, 2014, 34(10): 2725-2736. |
| [He Yuanhao, Zhou Guoying, Wang Shengjie, et al. Fungal diversity in Cunninghamia lanceolate plantation soil[J]. Acta Ecologica Sinica, 2014, 34(10): 2725-2736. ] | |
| [40] |
Yelle D J, Ralph J, Lu F, et al. Evidence for cleavage of lignin by a brown rot basidiomycete[J]. Environmental Microbiology, 2008, 10(7): 1844-1849.
doi: 10.1111/j.1462-2920.2008.01605.x pmid: 18363712 |
| [41] |
Frey S-D, Knorr M, Parrent J L, et al. Chronic nitrogen enrichment affects the structure and function of the soil microbial community in temperate hardwood and pine forests[J]. Forest Ecology and Management, 2004, 196(1): 159-171.
doi: 10.1016/j.foreco.2004.03.018 |
| [42] | 龚骏, 邢贝贝, 张倩倩. 隐真菌的研究进展[J]. 中国海洋大学学报(自然科学版), 2013, 43(11): 27-34. |
| [Gong Jun, Xing Beibei, Zhang Qianqian. Researching progress of cryptomycota[J]. Journal of Ocean University of China (Natural Science Edition), 2013, 43(11): 27-34. ] | |
| [43] | 刘德胜. 黄河三角洲盐碱地真菌多样性及活性次级代谢产物的初步研究[D]. 山东: 中国海洋大学, 2014. |
| [Liu Desheng. Preliminary Study on Diversity of Fungi Derived from Coastal Saline Soil in Yellow River Delta and Active Secondary Metabolites of Five Fungi[D]. Shandong: Ocean University of China, 2014. ] | |
| [44] | 丁翠. 酸性矿山废水污染胁迫下稻田土壤微生物菌群的演替[D]. 广州: 华南理工大学, 2019. |
| [Ding Cui. Responses of Microbial Communities to Intrusion of Acid Mine Drainage in Paddy Soils[D]. Guangzhou: South China University of Technology, 2019. ] | |
| [45] | 罗倩, 黄宝灵, 唐治喜, 等. 新疆盐渍土3种植被类型土壤微生物碳源利用[J]. 应用与环境生物学报, 2013, 19(1): 96-104. |
| [Luo Qian, Huang Baoling, Tang Zhixi, et al. Carbon source utilization of microbes in saline soil of three vegetation types in Xinjiang, China[J]. Journal of Applied and Environmental Biology, 2013, 19(1): 96-104. ] | |
| [46] | 许修宏, 成利军, 许本姝, 等. 基于高通量测序分析牛粪堆肥中细菌群落动态变化[J]. 东北农业大学学报, 2018, 49(3): 10-20. |
| [Xu Xiuhong, Cheng Lijun, XU Benshu, et al. Analysis of bacterial community dynamics in cow manure composting using high throughput sequencing[J]. Journal of Northeast Agricultural University, 2018, 49(3): 10-20. ] | |
| [47] |
王光华, 刘俊杰, 于镇华, 等. 土壤酸杆菌门细菌生态学研究进展[J]. 生物技术通报, 2016, 32(2): 14-20.
doi: 10.13560/j.cnki.biotech.bull.1985.2016.02.002 |
|
[Wang Guanghua, Liu Junjie, Yu Zhenhua, et al. Research progress of acidobacteria ecology in soils[J]. Biotechnology Bulletin, 2016, 32(2): 14-20. ]
doi: 10.13560/j.cnki.biotech.bull.1985.2016.02.002 |
|
| [48] |
Navarrete A A, Kuramae E E, Hollander, et al. Acidobacterial community responses to agricultural management of soybean in Amazon forest soils[J]. FEMS Microbiology Ecology, 2013, 83(3): 607-621.
doi: 10.1111/1574-6941.12018 pmid: 23013447 |
| [49] | 赵娇, 谢慧君, 张建. 黄河三角洲盐碱土根际微环境的微生物多样性及理化性质分析[J]. 环境科学, 2020, 41(3): 1449-1455. |
| [Zhao Jiao, Xie Huijun, Zhang Jian. Microbial diversity and physicochemical properties of rhizosphere microenvironment in saline-alkali soils of the Yellow River Delta[J]. Environmental Science, 2020, 41(3): 1449-1455. ] |
| [1] | 张彬, 郑新军, 王玉刚, 唐立松, 李彦, 杜澜, 田胜川. 1990—2022年天山北坡地区不同开垦年限耕层土壤盐分变化[J]. 干旱区研究, 2024, 41(9): 1435-1445. |
| [2] | 邱春霞, 刘晓宏, 李豆, 张佳淼, 李朋飞. 机载LiDAR和模糊推理系统在黄土高原土壤侵蚀监测中的应用[J]. 干旱区研究, 2024, 41(8): 1331-1342. |
| [3] | 万佳怡, 矢佳昱, 张华敏, 李兰晖, 丁明军. 三江源区不同覆被类型高寒草甸土壤水分变化特征[J]. 干旱区研究, 2024, 41(8): 1343-1353. |
| [4] | 董鹏, 任悦, 高广磊, 丁国栋, 张英. 呼伦贝尔沙地樟子松枯落物和土壤碳、氮、磷化学计量特征[J]. 干旱区研究, 2024, 41(8): 1354-1363. |
| [5] | 张培豪, 邢光延, 赵吉美, 刘昌义, 胡夏嵩. 轻度放牧和禁牧草地土壤物理力学性质特征——以夏藏滩滑坡区为例[J]. 干旱区研究, 2024, 41(8): 1364-1372. |
| [6] | 龙威夷, 施建飞, 李双媛, 孙金金, 王玉刚. 流域绿洲土壤盐分多模型反演效果评估[J]. 干旱区研究, 2024, 41(7): 1120-1130. |
| [7] | 郑柳娜, 江红南, 孙梦婷. 基于遥感影像的新疆渭干河—库车河三角洲土壤水盐与植被覆盖度的关系[J]. 干旱区研究, 2024, 41(7): 1131-1139. |
| [8] | 崔国龙, 李强峰, 高英, 刘维军, 张梅. 青海大通北川河源区典型植被土壤微生物群落结构特征及影响因素[J]. 干旱区研究, 2024, 41(7): 1195-1206. |
| [9] | 唐维春, 刘小娥, 苏世平, 田晓娟, 唐庆童, 张婧. 甘肃兴隆山不同演替阶段群落土壤氮素矿化对温度的响应[J]. 干旱区研究, 2024, 41(6): 984-997. |
| [10] | 毛光锐, 赵锦梅, 朱恭, 崔海明, 刘万智. 黄土高原高速公路边坡草本群落植被特征及其与土壤的关系[J]. 干旱区研究, 2024, 41(5): 788-796. |
| [11] | 雷菲亚, 李小双, 陶冶, 尹本丰, 荣晓莹, 张静, 陆永兴, 郭星, 周晓兵, 张元明. 西北干旱区藓类结皮覆盖下土壤多功能性特征及影响因子[J]. 干旱区研究, 2024, 41(5): 812-820. |
| [12] | 杨竹青, 王磊, 张雪, 申建香, 张伊婧, 李欣宇, 张波, 牛金帅. 典型固沙植物种子萌发和幼苗生长对土壤水分的响应[J]. 干旱区研究, 2024, 41(5): 830-842. |
| [13] | 洪国军, 谢俊博, 张灵, 范振岐, 喻彩丽, 付仙兵, 李旭. 基于多光谱影像的阿拉尔垦区棉田土壤盐分反演[J]. 干旱区研究, 2024, 41(5): 894-904. |
| [14] | 胡广录, 刘鹏, 李嘉楠, 陶虎, 周成乾. 黑河中游绿洲边缘三种景观类型土壤水分动态特征及影响因素[J]. 干旱区研究, 2024, 41(4): 550-565. |
| [15] | 张华, 押海廷, 徐存刚. 兰州市南北两山土壤水分遥感反演及植被需水量估算[J]. 干旱区研究, 2024, 41(4): 566-580. |
|
||